|
PULSE Research
Atomic and Molecular Dynamics
Ultrafast Chemistry
I3- in polymer matrix 
Figure 3 Typical WAXS image of I3- doped to 333mM concentration in a stretched PVDF polymer.
In this study, we will combine ultrafast x-ray measurements with femtosecond pump-probe spectroscopy of I3- embedded in a polymer matrix. The tri-iodide molecule is a prototypical reactive system for photoinduced fragmentation. I3- is particularly interesting because its gas and condensed phase photofragmentation differ dramatically. This makes the system well-suited for a study of the effects of the environment in photochemistry. For the case of x-ray diffraction, I3- has several advantages. Low frequency motions are well-suited to the anticipated x-ray pulse durations. A large number of electrons in the iodine atomic core enhances the x-ray scattering signal and provides a high contrast between the chromophore and the surrounding polymer matrix. Finally, this system provides an opportunity for a high density of isolated and aligned chromophores, in which alignment is achieved simply by stretching the polymer. We recently enjoyed a very successful x-ray scattering beam time on the SSRL beam-line 11.3. This was a three day run on a WAXS machine during which we investigated both liquid and polymer solutions of the I3- anion in preparation for hard x-ray scattering of polymer based aligned molecular systems. Ultrafast X-ray Spectroscopic Studies of Excited State Electronic Structure The initial stages of efficient photochemical reactions invariably occur on the femtosecond to picosecond time scale, as exemplified by bacterial photosynthesis and the bacteriorhodopsin proton pump. Identifying the mechanisms for efficiently channeling the energy of a photon to a desired chemical product will be the overarching objective of the research. This objective will be pursued by studying the chemical dynamics of photocatalysts that store solar energy via photo-initiated electron transfer reactions. The emphasis will be placed on organo-metallic electron transfer chemistry because of the strong visible absorption bands in numerous organo-metallic compounds and the diverse catalytic power of multinuclear metal centers as demonstrated by a variety of metallo-enzymes. Ultrafast electronic spectroscopy with near-IR, visible, and ultraviolet radiation has been widely utilized to investigate photo-induced dynamics in molecular systems, but for transition metal organo-metallic systems, the electronic structure has sufficient complexity that the interpretation of ultrafast measurements of electronic dynamics generally prove inconclusive. X-ray spectroscopy has many attributes that enhance the interpretive power of the measurement. With ultrafast x-ray spectroscopic measurements, we will be able to harness these strengths and utilize them to further our understanding of excited state electronic structure and dynamics. Atomic-resolution mapping of excited electronic structure with ultrafast soft x-ray spectroscopy: We have begun the development of ultrafast soft x-ray spectroscopy as a probe of excited electronic structure in photo-voltaic and photoelectrochemical materials. Our initial studies have investigated the excited state properties of cuprous oxide thin films, which we have grown in collaboration with the Environmental Molecular Sciences Laboratory at PNNL. We measured the change in both the Cu L3-edge and the O K-edge absorption induced by laser excitation with 400 nm light. By measuring the change in absorption for both the Cu L3-edge and the O K-edge, we can map the changes in electronic structure with atomic specificity, as shown in the Fig. 1. 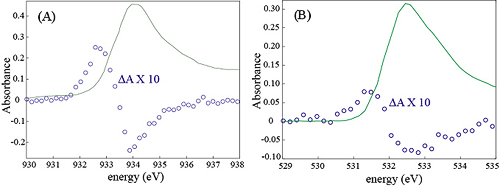
Fig. 1: Changes in the Cu L3-edge and the O K-edge induced by electron-hole pair creation with 400 nm laser light in a cuprous oxide thin film. The change in absorption for the copper edge exceeds that of the O K-edge by roughly a factor of 2, indicative of the laser induced changes in the charge distribution.
Resonant inelastic x-ray scattering (RIXS) studies of charge, spin, and covalency dynamics: X-ray spectroscopy measurements at the at the LCLS x-ray free electron laser being built at SLAC will be very different than spectroscopy measurements at current synchrotron facilities. In particular scanning the x-ray energy over hundreds to thousands of eV will not routine. Hard x-ray emission will be the simplest spectroscopic technique to implement from the source perspective, since it only requires the x-ray source to excite a sample above the chosen x-ray absorption edge. Once the lasing properties of LCLS have been established, tuning the x-ray energy to a given atomic edge within the energy range of the LCLS should be achievable and will make resonant inelastic x-ray scattering (RIXS) measurements possible. The x-ray emission that results from the 3p electron filling of a 1s core hole provides a sensitive probe of the charge and spin on the x-ray emitting atom, in particular for manganese and iron compounds. We will utilize this attribute of x-ray emission spectroscopy (XES) to probe the time evolution of charge and spin degrees of freedom in laser excited Mn and Fe complexes. To enable the development of ultrafast XES and RIXS in our research group, we have been acquiring the experimental and theoretical training required to conduct and comprehend steady state XES and RIXS measurements. We have chosen solid state RbMnFe(CN)6, manganese Prussian blue, as a model photo-induced electron transfer system for developing time resolved XES spectroscopy. RbMnFe(CN)6 undergoes a thermal phase transition from the Mn2+(S=5/2)-NC-Fe3+(S=1/2) cubic high temperature (HT) phase to the Mn3+(S=2)-NC-Fe2+(S=0) tetragonal low temperature (LT) phase caused by the electron transfer between Mn to Fe and a Jahn-Teller distortion around the Mn3+ ion. The low temperature phase can also be transiently photo-excited to the high temperature phase well below the phase transition temperature. This photo-induced phase transition has been attributed to a metal-to-metal charge transfer from Fe2+ to Mn3+. The XES spectra for the high and low temperature phases appear in Fig. 2. 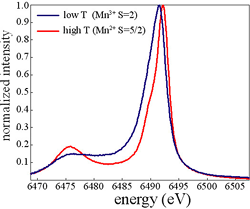
Fig. 2: X-ray emission spectra in the manganese Kβ1,3 region for the high and low temperature phases of manganese Prussian blue.
We have chosen a solid state photoinduced phase transition sytem, rather than a solution phase chemical system, to investigate initially because it will optimize the concentration of excited states and consequently the transient changes in the XES spectrum. At the LCLS, where the laser coincident x-ray flux greatly exceeded that achievable at current synchrotrons, solution phase studies will be possible and we will be able to observe the time evolution of the spin multiplicity of photon induced spin crossover complexes. 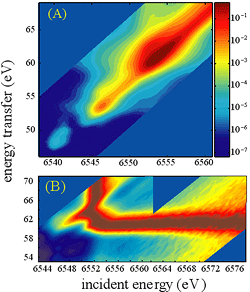
Fig. 3: (A) Logarithmic intensity RIXS spectrum for the manganese K-edge measured in the Kβ1,3 region. (B) Normalized RIXS spectrum, with the peak in the absorption spectrum for each emission energy being normalized to one. This clearly shows the prominence of a Raman feature with an energy transfer of 61 eV. This corresponds to the energy difference between the K-edge main peak and the Kβ1,3 peak.
We have initiated steady state XES and RIXS investigations of the electronic structure in this compound to study the thermal phase transition, and will be initiating optical transient absorption measurements following the commissioning of the transient absorption component of our laser laboratory. A steady state RIXS spectrum for the high temperature phase can be found in Fig. 3. Following these initial optical studies, we will conduct time resolved x-ray absorption, x-ray emission, and powder diffraction studies at the Advance Photon Source. By combining time resolved powder diffraction with x-ray spectroscopy, we have the potential to independently track the structural and electronic degrees of freedom, making it possible to investigate their interdependence. Multidimensional Vibrational Spectroscopy Studies of Thermal Reactions in Solution Fast thermal reactions can be investigated with molecular dynamics simulations, providing detailed atomic scale information about reaction mechanisms, but experimental techniques capable of corroborating simulation results have been lacking. While multi-dimensional NMR provides a powerful technique for studying kinetics, it lacks the necessary femtosecond resolution for investigating dynamics. Multi-dimensional vibrational spectroscopy provides an analogous technique with the necessary time resolution to study the fast conformational dynamics accessible to molecular dynamics simulations. Hydrogen bond dynamics in ionic solutions: Aqueous electrolyte solutions lubricate the chemical machinery of natural and biological systems. While the unique and incompletely understood properties of water receive significant and justified attention, natural and biological processes also depend critically on the ionic species present in solution. Some studies have concluded that the structure and dynamics of water outside the first solvation shell of the ions in solution remain unaffected by the presence of ions in solution, and electrolyte solutions can be considered as a mix of two water ensembles, one residing in the first ionic solvation shell and the other ensemble ostensibly unaffected by the presence of ions in solution. While this depiction cannot account for all aspects of the electrolyte solution structure observed in experiment, the extent to which these structural effects can be disregarded with respect to the dynamics of aqueous ionic solutions remains unresolved. To understand the properties of ionic solutions requires not only a detailed understanding of the dynamics and structure of these proposed populations, but also a full understanding the inter-conversion mechanism and dynamics for these two hypothesized populations of water molecules. While the lifetime of a molecule in the first solvation shell can be determined robustly in a molecular dynamics simulation, the lifetime has never been measured directly in an experiment. 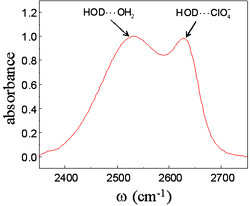
Fig. 4: Vibrational absorption spectrum of the deuterated hydroxyl stretch of isotopically mixed water in an aqueous 6M sodium perchlorate solution. The peak at low frequency corresponds to OD stretches that donate hydrogen bonds to other water molecules while the peak at high frequency corresponds to OD stretches that donate hydrogen bonds to perchlorate anions.
We have used multi-dimensional vibrational correlation spectroscopy (MVCS) to determine the lifetime of a H-bond between a water molecule and a perchlorate ion, as well as the spectral diffusion dynamics for each water configuration. The hydroxyl stretch vibrational absorption peak for hydroxyl groups donating a hydrogen bond to a perchlorate anion results in an absorption peak distinct from hydroxyl groups donating a hydrogen bond to another water molecule, as shown in Fig. 4. We have used multi-dimensional vibrational correlation spectroscopy to monitor the thermally induced interchange of these two hydrogen bond populations, by monitoring the growth in the off-diagonal signal in the correlation spectra, as shown in Fig. 5. These measurements, in conjunction with time resolved population and anisotropy measurements, provide a very detailed picture of the dynamics in ionic solution. 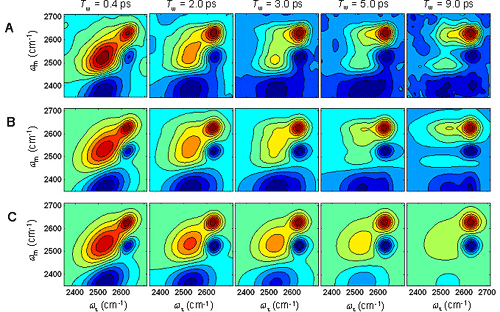
Fig. 5: 2D vibrational correlation spectra of the OD stretch in an isotopically mixed aqueous solution of 6M sodium perchlorate collected at a series of waiting times, Tw. The peak at higher frequency, 2633 cm-1, corresponds to OD groups that donate a hydrogen bond to a perchlorate ion, while the peak at lower frequency, 2534 cm-1, corresponds to OD groups that donate a hydrogen bond to another water molecule peak. The peaks in red along the diagonal correspond to OD groups that reside in the same general hydrogen bonding configuration, the peaks in blue below the diagonal result from vibrational excited state absorption, and the intensity in the upper left quadrant of the correlation spectra results from OD stretches that initially donated a hydrogen bond to a water molecule switching to donation to a perchlorate anion. The experimental data is in (A) the simulation of the experiment appears in (B), and the simulation without exchange appears in (C). The H-bond exchange time is 17±4 ps.
Mechanistic Studies of First-Order Phase Transitions Using Ultrafast X-ray Scattering First-order phase transitions are activated processes where a free energy barrier impedes the transition from one phase to another [1]. Understanding the mechanism of 1st-order phase transitions requires the determination and characterization of the atomic and molecular motions involved in crossing this activation barrier [2]. While x-ray and neutron inelastic scattering access the equilibrium structure and dynamics characteristic of stable phases, they do not interrogate the ultrafast, but rare, barrier traversal events. Ultrafast laser excitation provides a means of depositing energy with sufficient magnitude and rapidity that the dynamics of the phase transition can be accessed directly in the time domain [3-6]. The rate of ultrafast laser induced phase transitions have been resolved with a variety of experimental probes, including transient optical absorption and reflectivity,[6-9] as well as time resolved x-ray scattering [3-5,10,11]. These time resolve ultrafast x-ray scattering measurements of laser melting highlight the power of non-equilibrium time domain techniques and the current limitations of our understanding. Dynamics of photo-induced melting in semiconductors: We performed a series of ultrafast x-ray scattering experiments at the Sub-Picosecond Pulse Source (SPPS) that utilized the SLAC linear accelerator to generate 80 femtosecond duration hard x-ray pulses. These studies investigated the influence of carrier density on the stability of crystalline InSb, a tetrahedrally bonded semiconductor with the same crystal structure as GaAs. The laser disordering and melting of semiconductors represents a classic demonstration of the limitations of optical studies and the potential of ultrafast x-ray sources for studying structural dynamics. Early experimental work demonstrated that intense fs pulse excitation of a silicon crystal led to a sub-ps increase in the reflectivity. Although absorption of the laser generates an increased reflectivity due to the direct excitation of carriers, the magnitude of the observed change could not be explained by this effect alone and led to the conclusion that intense laser excitation causes a semiconductor to metal transition. Since Si forms a metallic liquid, metal formation was attributed to crystal melting. The rate of this purported phase transition exceeds the rate of energy transfer from the excited electrons to the crystal vibrations, leading to the supposition that the melting occurs non-thermally. Theoretical studies have supported this supposition by predicting that excitation of roughly half an electron per atom generates a crystal instability that spontaneously leads to crystal melting. Even though ultrafast changes in the visible reflectivity led to this hypothetical non-thermal melting mechanism, these optical studies cannot unambiguously confirm the conjecture. We performed a series of studies of laser induced melting in InSb to determine whether melting occurred due to an electronically generated crystal instability or due to thermal excitation of large amplitude vibrational motion. In our studies, we observed three disordering regimes. At the lower laser fluences, the diffraction intensity decayed exponentially, consistent with thermal disordering where the exponential time constant reflected the rate of phonon emission by the laser excited carriers. For a wide range of laser fluence, the initial decay in diffraction intensity displayed a Gaussian time profile with a time constant of 400 femtoseconds. This time constant and the Gaussian time profile provided strong evidence that the constant velocity inertial disordering dominated the decay in the diffraction intensity. For the shallowest x-ray probe depths and highest laser fluence, we observed accelerated disordering, as predicted by theory. 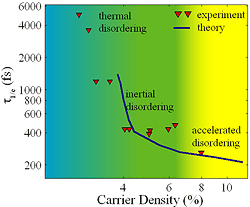
Fig. 6: Comparison of experimental and theoretical decay times for the (111) Bragg peak of InSb. In the experiment the decay results from laser excitation of electron-hole pairs, while the theory models laser excitation with an elevated electronic temperature. The qualitative agreement between theory and experiment confirms that carrier generated changes in the potential energy surface dictate the initial rate of disordering in InSb.
To confirm that the observed dynamics at high carrier densities did result from changes in the potential energy surface, we calculated the carrier density dependence of the phonon dispersion curves of InSb with density functional theory calculations. These calculations use an elevated electronic temperature to simulate the effect of laser excitation, by filling the calculated valence and conduction bands with the Fermi-Dirac distribution. We then used a modified Debye-Waller model and the calculated phonon dispersion curves to calculate the time dependent diffraction experiment. To compare the results of theory and experiment, we parameterized the time dependent diffraction intensities with the 1/e time (the time required for the initial diffraction intensity to drop by a factor of 1/e). The comparison between experiment and theory appears in Fig. 6. The qualitative agreement between theory and experiment demonstrates that the initial disordering dynamics are dominated by changes in the potential energy surface generated by electronic excitation, not by thermal excitation of large amplitude vibrational motion. 
|